Checking for PCR duplication problems, contamination, etc.
Checking for levels of PCR duplication
Any protocol that uses a PCR step can have problems with PCR duplication. If one does paired-end sequencing, the fragment end position may allow us to filter PCR duplicates out. For that you can use tools such as Picard tools’ MarkDuplicates. However, if you cannot do that because you did single-end sequencing, PCR duplicates may be a problem. The usual expectation is that different reads covering a site stem from different cells. However, if some of the reads actually go back to the same fragment that was amplified by PCR, the reads are not independent samples. If a genotype has 20 reads that all support the same allele, one would be certain that it is homozygous. Missing the second allele with 15 reads is unlikely. However, if in fact the 15 reads stem from the same original DNA fragment that was amplified by PCR, i.e. the reads represent different PCR duplicates, missing the second allele is highly likely. We cannot trust genotypes with high PCR duplication levels.
If levels of PCR duplication are low, we expect that with increasing numbers of reads for a given individual, more and more loci are sequenced as the reads are distributed randomly among loci. At some threshold of number of reads all loci should be present in an individual. Adding more reads will only increase sequencing depth but not number of loci as all loci are already sequenced. If levels of PCR duplication are very high, the number of loci does not increase as quickly with the number of reads as expected. This is because the reads are not random samples of the different loci but are pseudo-replicates of a limited number of starting fragments.
As explained in the lecture, we can plot the number of reads against the number of RAD loci. Note, this does not work with whole-genome data. Joana Meier wrote a script to do this on RAD data.
You can run it without any arguments in your folder where you have the bam files. It will generate a mapping report which contains the number of reads and RAD loci found in each individual.
createRADmappingReport.sh
The mapping report has the following columns: sample sampleLib mappedReads lociN meanDepth lociNmin10reads meanDepthMin10reads
As I call the samples as 123.PunNyer.lib1, the first column will give the sample name (123.PunNyer) and the second column gives the full name. If you have a different naming scheme without “.” in the sample name, the two columns will likely be the same. The next column gives you the number of mapped reads per individual, then the number of RAD loci sequenced, and the mean depth at all of these loci. The last two columns give the number of loci with at least 10 reads and the mean depth at those loci.
I would then plot for each individual the number of loci with at least 10 reads against the number of reads:
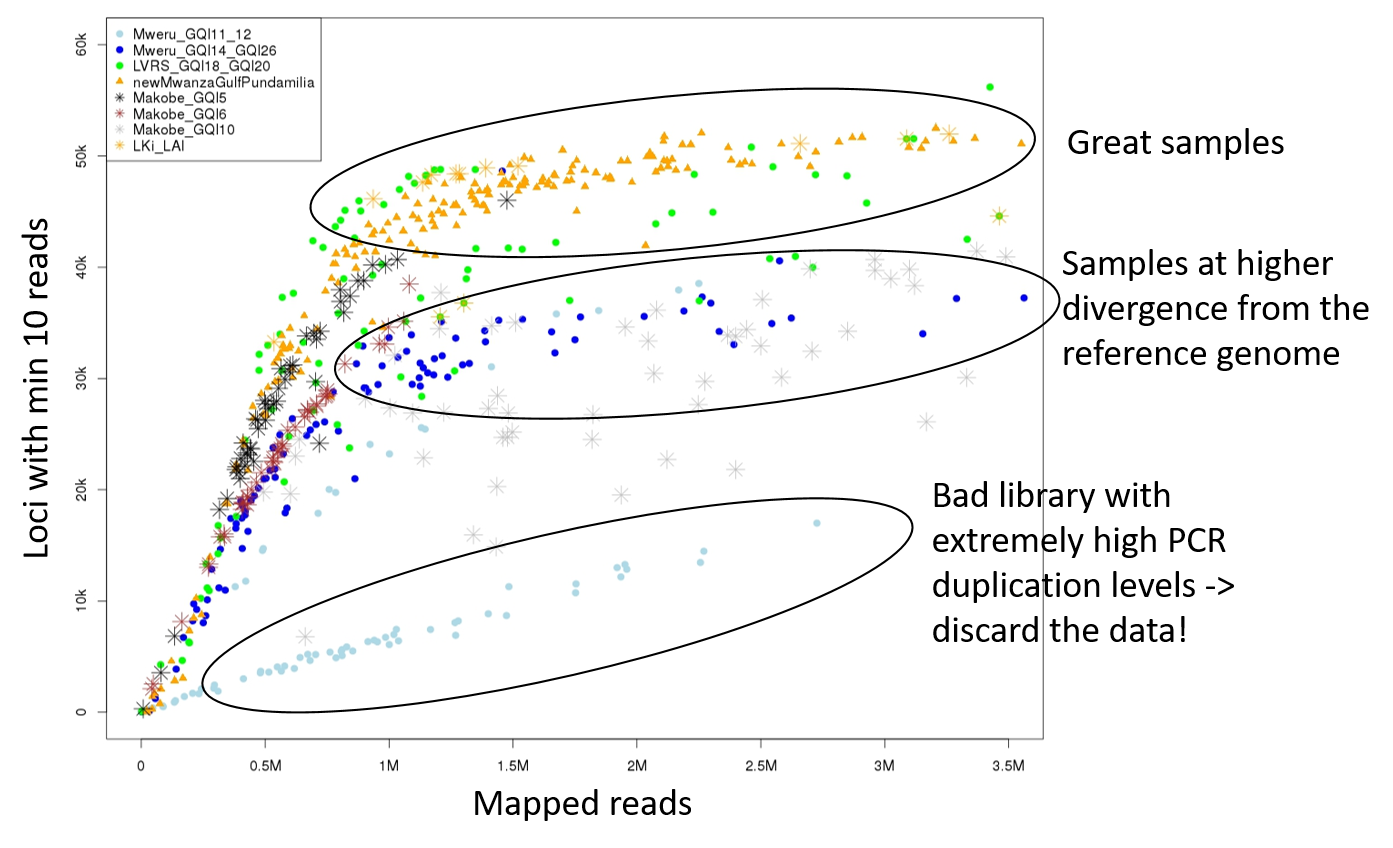
In this example figure, each dot is an individual and the colour and symbol indicates to which library it belongs. Some libraries performed very well which can be seen as a fast increase of number of loci with number of reads. At about 1 million reads, most individuals reach the total number of RAD loci and therefore the number of loci does not increase anymore with increasing number of reads. However, an individual in the bad libraries highlighted on the bottom with 1 million reads has way less loci. This is because a large proportion of the reads represent PCR duplicates of only a few DNA fragments. The intermediate circle shows individuals that are well-sequenced but they have less loci because they are divergent from the reference genome. Some of their RAD loci are too divergent to map to the reference and are thus missing. If these samples were equally divergent from the reference as the other samples, their position in the plot would indicate high PCR duplication levels.
Using allelic balance at heterozygotes to detect contamination
Heterozygote positions can be very useful to detect contamination, high PCR error prevalence, Illumina index switching or other issues generating erroneous heterozygotes such as paralogous regions. Without PCR duplicates and other issues, the reads at any genotype should reflect independent copies of the maternal and paternally inherited genes.
At heterozygote genotypes, the two alleles should thus be represented with equal number of reads (except for stochastic differences). However, if some of the reads represent PCR duplicates the variance in the number of reads supporting each allele (allelic balance). PCR errors present in multiple PCR duplicates can cause erroneous heterozygote genotypes. If the heterozygote genotype is due to such a PCR error or to contamination or Illumina index switching, the erroneous reads are expected to be rarer than the correct reads. Such heterozygotes should thus show a strong allelic disbalance, whereby the ratio of reads supporting each allele deviates from 50% even at high sequencing depth.
If cross-contamination among samples due to lab errors or Illumina index switching is the main reason for heterozygotes with uneven read counts, we would expect singletons (i.e. sites where only a single individual carries an alternative allele) to show better allelic balance than positions with higher minor allele frequency across all samples. This is because the reads supporting the underrepresented allele come from another individual should also be found in that individual and can thus not be singletons in the dataset.
In contrast, if PCR errors present in multiple PCR duplicates are the main reason, singletons should show the strongest signal. If highly unbalanced genotypes are due to contamination by another individual in the dataset or due to Illumina barcode switching, singletons (i.e. positions in the genome with only a single individual carrying an alternative allele) are expected to show better allelic balance than positions with higher allele frequency in the dataset.
This is because if the reads supporting the underrepresented allele come from another individual, they should also be found in that individual and thus not be singletons in the dataset. However, if the unbalanced heterozygotes are due to PCR errors in PCR duplicates, they should be most pronounced in singletons. This is because when PCR errors are randomly distributed across the genome, they are most likely to hit a monomorphic site because monomorphic sites are most frequent in the genome.
To check the heterozygotes, you can use the script originally written by David Marques and apply it on a dataset of SNPs. It takes a SNPs-only vcf file as input. It generates a plot for each individual, revealing potential contamination. Let’s try it on a few samples that were RAD-sequenced:
FILE=Pundamilia.RAD.subset
checkHetsIndvVCF.sh $FILE.vcf.gz
This script will generate an output called $FILE.hetIndStats.pdf that visualizes the allelic balance at heterozygote positions in a separate page for each individual. This file can then be used to detect contaminated individuals or barcode switching problems.
Here an example of a well-sequenced individual without evidence of contamination:
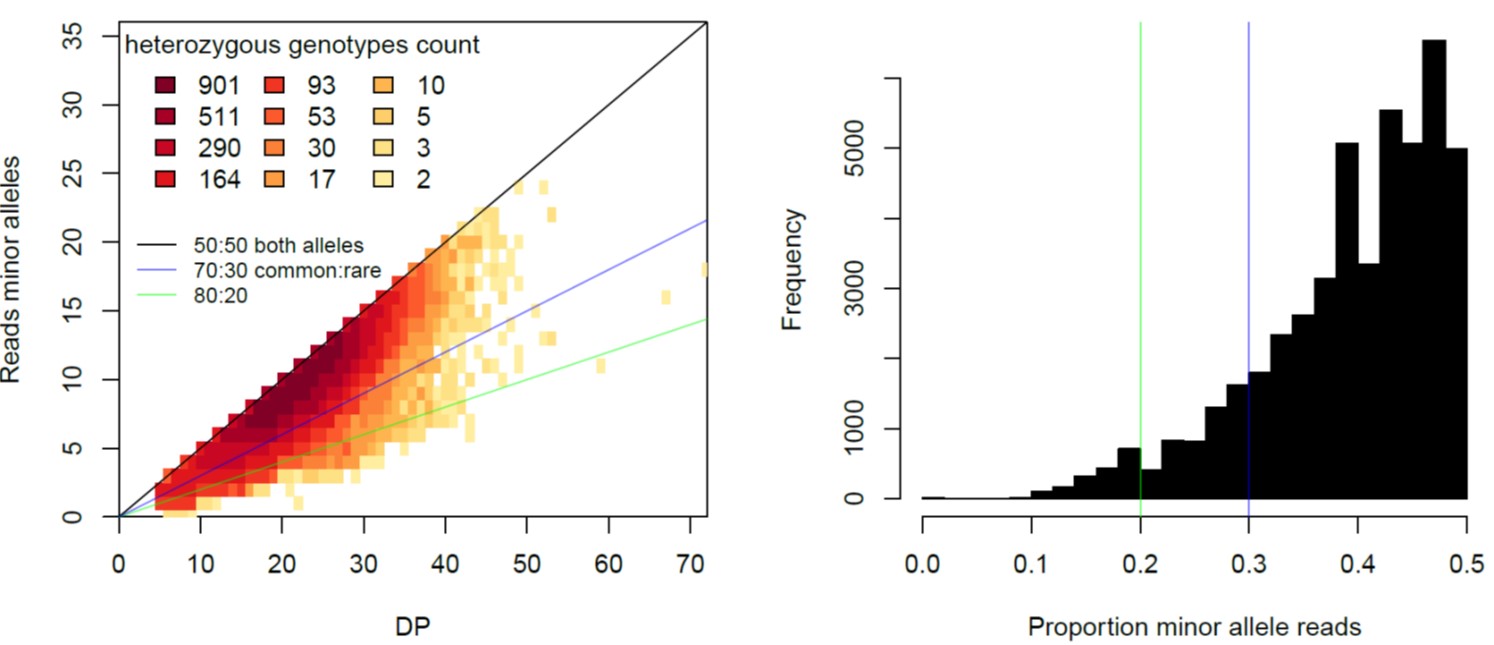
The plot on the left shows that most heterozygous positions have more or less equal number of reads for both alleles. The plot on the right shows the distribution of percentage of the proportion of the reads for the minor allele (i.e. the allele that has less reads in that individuals).
Here an example of a badly contaminated individual
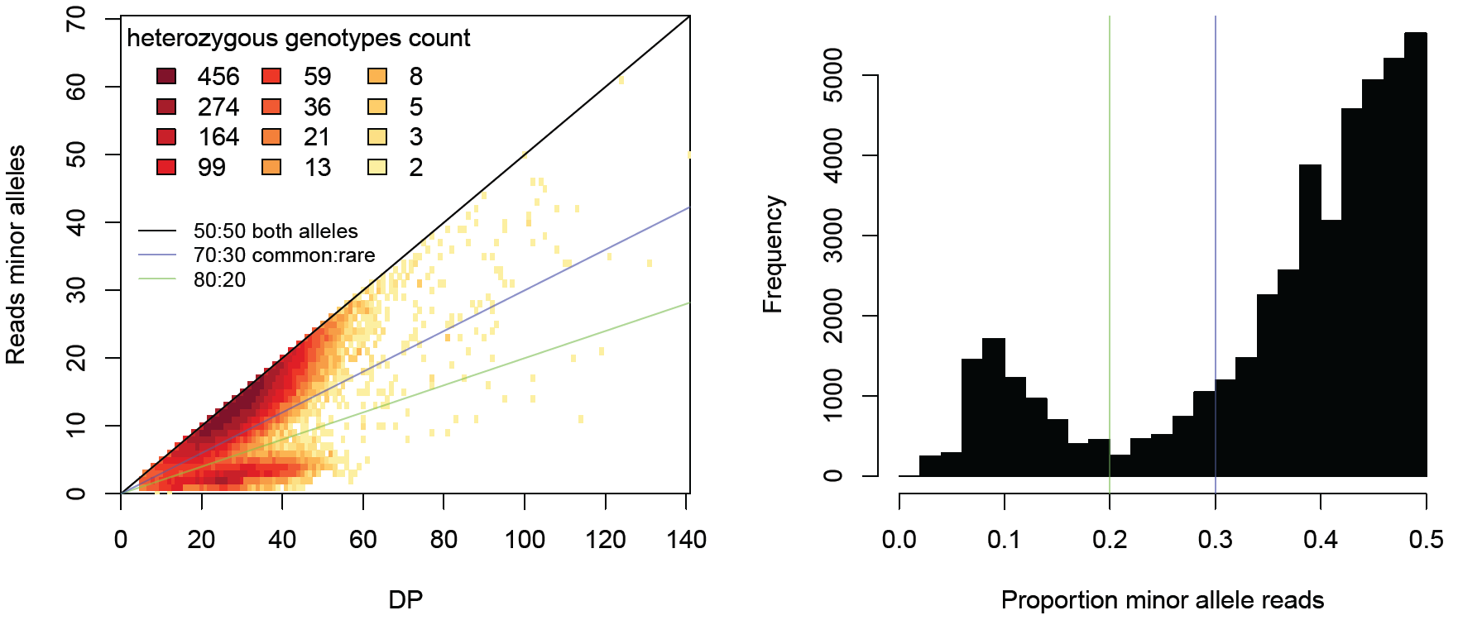
You can see that many heterozygote sites one allele is covered by much less reads than the other allele. These heterozygote positions with strong imbalance of reads supporting the two alleles indicate contamination. Alternatively, if you are using a PCR-based library preparation method (e.g. RADseq), PCR duplicates may cause large variation in the number of reads supporting each allele. We would expect both alleles to be represented by more or less equal numbers of reads, particularly if sequencing depth is high. However, if multiple reads actually stem from the same PCR fragment, the variance in number of reads per allele increases and can be highly biased if some PCR fragments are smaller and thus amplified more during the PCR. In addition, the PCR enzymes make mistakes and PCR errors will look like alleles supported by fewer reads than the real allele. For whole-genome resequencing data, PCR duplicates can be removed, but if you have ddRAD data or single-end sequenced RAD data, the PCR duplicates can cause unbalanced heterozygotes.
To distinguish if strongly unbalanced read counts are due to cross-contamination or PCR errors in PCR duplicates, we will extracts singleton sites and run the script a second time on this subset:
vcftools --gzvcf $FILE.vcf.gz --max-mac 1 --mac 1 --recode --stdout | gzip > $FILE.mac1.vcf.gz
checkHetsIndvVCF.sh $FILE.mac1.vcf.gz
If the sites with highly unbalanced read counts disappear, this is evidence for PCR errors and against cross-contamination.
Fixing low-level contamination
If there is evidence for low levels of contamination, it is possible to call genotypes with GATK HaplotypeCaller specifying –contamination_fraction_to_filter. Alternatively, heterozygote genotypes with highly unbalanced read counts can be filtered out e.g. with the script of Joana Meier.